
Fact from 2,500 of FDA Form 483s
This article is a translation of a Japanese article published on 「PHARM TECH JAPAN Vol.41 NO.6(2025)」.
Introduction
The FDA conducted 194 inspections of domestic drug manufacturing facilities, including API manufacturing sites from 2018 to 2024, and made findings in 138 inspections. Of those 138 inspections, data integrity issues were found in 81 inspections. In addition, the revised Japan GMP Ministerial Ordinance in 2021 has set out requirements on data integrity, so complying with data integrity requirements is an urgent task.
It is said that the requirements of Data integrity (DI) are ALCOA, however, even if you read various guidance on DI and delve deeply into our head for ALCOA, it is difficult to reach the DI practice level expected by the authorities. The best way to reach the DI practice level expected by the authorities is to learn from findings of authorities’ inspections. The FDA is the most open authority in disclosing inspection findings, and all the inspection findings are available. This article introduces the trends of DI findings of over 2,500 FDA inspection observations from the perspective of record reliability and manufacturing control integrity.
1. What is data integrity?
Data integrity means that data are complete, consistent, and accurate; in other words, data integrity compliance means continuously ensuring the reliability of records throughout the retention period (see the article-by-article commentary of Article 8-2 of the GMP Ministerial Ordinance.
1.1 Requirements for data integrity compliance
Many DI guidance documents have been issued by regulatory authorities (see Table 1) and industry groups. According to these documents, the requirements for data integrity compliance are the ALCOA Principles and its extended version, ALCOA++ (see Table 2).
Table 1 DI guidance issued by regulatory authorities etc.
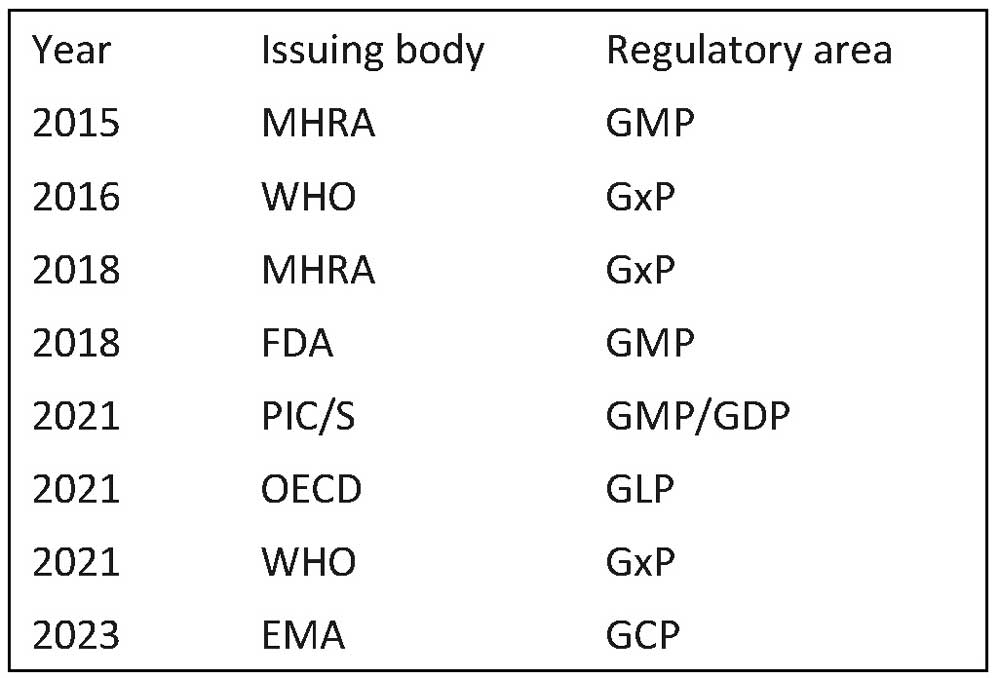
Table 2: ALCOA Principles and ALCOA++

1.2 Terminology related to data integrity
Here are some terms related to DI compliance you may see in guidance issued by regulatory authorities, etc.
● Raw Data
PIC/S GMP Definitions in "Chapter 4 Documentation":
At least, all data on which quality decisions are based should be defined as raw data.
Explanation in GMP 20-4 of Q&A on Japan GMP (2022 Edition)
Raw data must be capable of verifying that the results are correct. For example, the raw data related to testing and inspection include the following:
- Data printed from the test devices
- Chart output from the recorder or a record of the values
- Values written down by displayed on measuring device
- A record of observations
- A file electronically recording charts and other waveform data
- photograph
- Records of the process of calculations, conversions, etc., using the above data
From the perspective of data integrity, "raw data" should be stored so that it is available in the status it was obtained.
● Critical Process Parameter (CPP)
- Process parameters whose variation gives impact on Critical Quality Attributes (CQAs)
- Monitoring and management are required to ensure the required quality
● Metadata: Metadata is information needed to understand the meaning of data. Data alone does not mean anything if it is just a numerical value. For example, the number "23" is meaningless without metadata of units such as "mg". Metadata also includes timestamp of the time the data collected, the user ID of the person who performed the test or analysis generating the data, the ID of the equipment used to collect the data, and audit trails. Metadata is raw data.
● Original record: The first information captured on paper or electronically.
● True Copy: An exact verified copy of an original record.
● Dynamic Record: A record in a format that allows data processing, data analysis, and enlarged view of data. For example, an original record chromatography that is maintained electronically. If the original record is a dynamic electronic record, the original record or a true copy of it must be maintained electronically.
● Static Record: A record in a static format such as paper or PDF, where the data is fixed and does not allow reprocessing or enlarged view. Static records may be maintained in paper or PDF format. However, PIC/S Data Integrity Guidance for Inspectors states in section 7.7.2 that the following should be included in the data stored:
- Metadata
- Relevant audit trails
- Result Files
- Software/system configuration settings specific to each analytical run
- A complete record of all data processing steps required to reconstruct the raw data set, including methods and audit trails
- All data processing runs (including methods and audit trails) necessary for reconstructing the raw data set
However, it is stated that maintaining the above data on paper is onerous from the point of view of GMP. It seems to suggest that maintaining them electronically is easier than being on paper. In addition, maintaining electronic records of pH meters that are considered as static records is requested in an FDA inspection, and I will discuss this later.
1.3 Ensuring the authenticity of electronic records
ERES Guideline issued in 2005 lists the following 3 requirements for ensuring the authenticity of electronic records.
● security: To limit system access only to authorized people by IDs and passwords, etc., and prevent unauthorized changes or deletion of data.
● Audit Trail: Historical information on the creation, modification, deletion and manipulation of records by humans, including who, when, from what to what, and the reason for modification or deletion. It is the same concept as when modifying or deleting handwritten raw data.
● backup data: To protect against loss due to system failures, or other disasters. Backups should be protected from modification or damage so that it can be used for system recovery in case of emergency.
The above 3 requirements for ensuring the authenticity of electronic records can be considered as technical elements for ensuring data integrity in computerized systems.
1.4 Regulatory authorities' expectations on DI practices
Even if you understand the above DI terms, read many DI guidance, and delve deeply into ALCOA in your head, it is difficult to meet the expectation on the DI practice of inspectors who are familiar with various DI deficiencies. We should learn how to implement DI practice from deficiencies found in inspections by regulatory authorities. The FDA is the most open authority in disclosing inspection findings, and all the inspection deficiencies are available. This article introduces the trends of DI related deficiencies listed in over 2,500 FDA inspections findings that were obtained through paid disclosure based on the Freedom of Information Act (FOIA).
2. Disclosure of FDA Inspection Observations
When the FDA identifies non-compliance activities during an inspection of a pharmaceutical manufacturing site, the inspectors document these as "observations" on an FDA Form 483, which is issued to the site on the final day. The site must respond with corrective actions or the situation of corrections. If the response is inadequate, FDA may issue a Warning Letter (WL) to the manufacturing site. The manufacturing site must response to the WL and inappropriate response can lead to administrative actions, such as import alerts or recalls.
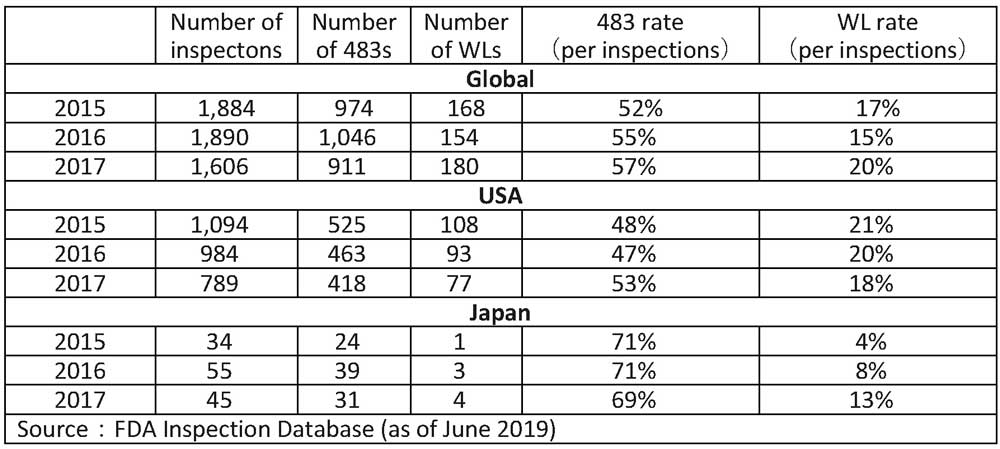
All WLs are published on the FDA's website; you can search WL by key words such as the name of the company. However, Form 483s can only be obtained through a fee-based disclosure request, and even a Japanese can also apply for disclosure. The number of 483s and WLs issued from 2015 to 2017 is shown in Table 1. The FDA issues around 1,000 Forms 483s annually, so the 2,500 Form 483s referred to in this article cover roughly 483s in 2.5 years.
A Form 483 lists all the observations made by all the inspectors, but WL only lists the observations to which the response is insufficient, and the FDA judges them to be non-compliant with regulations. It is a good method to find out the actual findings at the inspection site by collecting and sorting out the observations listed in Form 483.
Table 3: FDA GMP Inspection Annual Inspection, 483, and WL Numbers
About Author
Career The profile is the information as of the time of writing.
Related Articles
Search by Keywords


Comment
/
/
/
Comment