
はじめに
ここまで、生きた細胞を製品とする細胞製造性を考慮した再生医療等製品の製品設計の考え方と、それに付随して生じる製品の同等性を担保する製造再現性の考え方について、また具体的なお話しとして、製品の品質特性(CQA)と切り離すことの難しい原料等や細胞加工の操作など、工程設計について雑感を述べました。今回からは、これらの考え方を前提とした、運用設計(マネジメントシステム)についてお話しをしたいと考えております。それに伴い、本稿では、GCTP省令の復習をあらためて行わせていただき、また、昨年11月に厚労省の事務連絡で発出された「再生医療等製品の無菌製造法指針」について雑感を述べさせていただきます。
● あらためて、GCTP省令に書かれている内容を確認する
GCTP省令については、第5回において、GMP省令との違いについて概説しました。今回は、再生医療等製品固有の考え方を除外し、「製造できること」を前提として、あらためて、GCTP省令について整理してみましょう。GMP省令を理解している方には常識の範疇となる内容ですが、お付き合いいただければ幸いです。
GCTP省令(平成26年厚生労働省令第93号)は、第1条から第23条までの文章で構成されていますが、その構成は前述した通り、GMP省令と変わりません。第1~3条(趣旨、定義、適用範囲)を除くと、図のように、品質確保において最も重要となる、品質リスクマネジメント(祭4条)、製造部門及び品質部門(第5条)、製造管理者(第6条)および職員(第7条)の要求が示され、品質リスクマネジメントを含む、マネジメントシステムの構築についての要求が前提となります。ここで特に求められるのは、トップマネージメントによる組織体制と、適切な職員の充当となります。その上で、品質確保すべき製品を対象とした製品標準書(第8条)と手順書等(第9条)の準備について、文書化要求が行われています。製品標準書には、製品規格や原料等の基準、製造方法および手順、品質の試験方法および手順、製品の包装形態、表示材料および表示方法、保管条件、細胞加工作業者に要求されるスキルなどを全て記載すべきものであり、再現性よく製造が実施できることを前提としています。また、手順書等では、上位文書として、衛生管理基準書、製造管理基準書、品質管理基準書(三基準書)を必須とし、後述する出荷管理、バリデーション、品質照査、変更管理、逸脱管理、回収処理、自己点検、教育訓練、記録管理などに関する手順作成が要求されており、製造管理者は品質リスクマネジメントを活用することで、製品のライフサイクルを通して適切に製造管理および品質管理を運用できるようにする組織の構築が必須となります。実際に、GCTP調査に係るサブシステムの1丁目1番地は「組織」であり、その他の一番地は全て手順書・記録書であり、ここをクリアしていなければGCTP省令への対応はあり得ません。
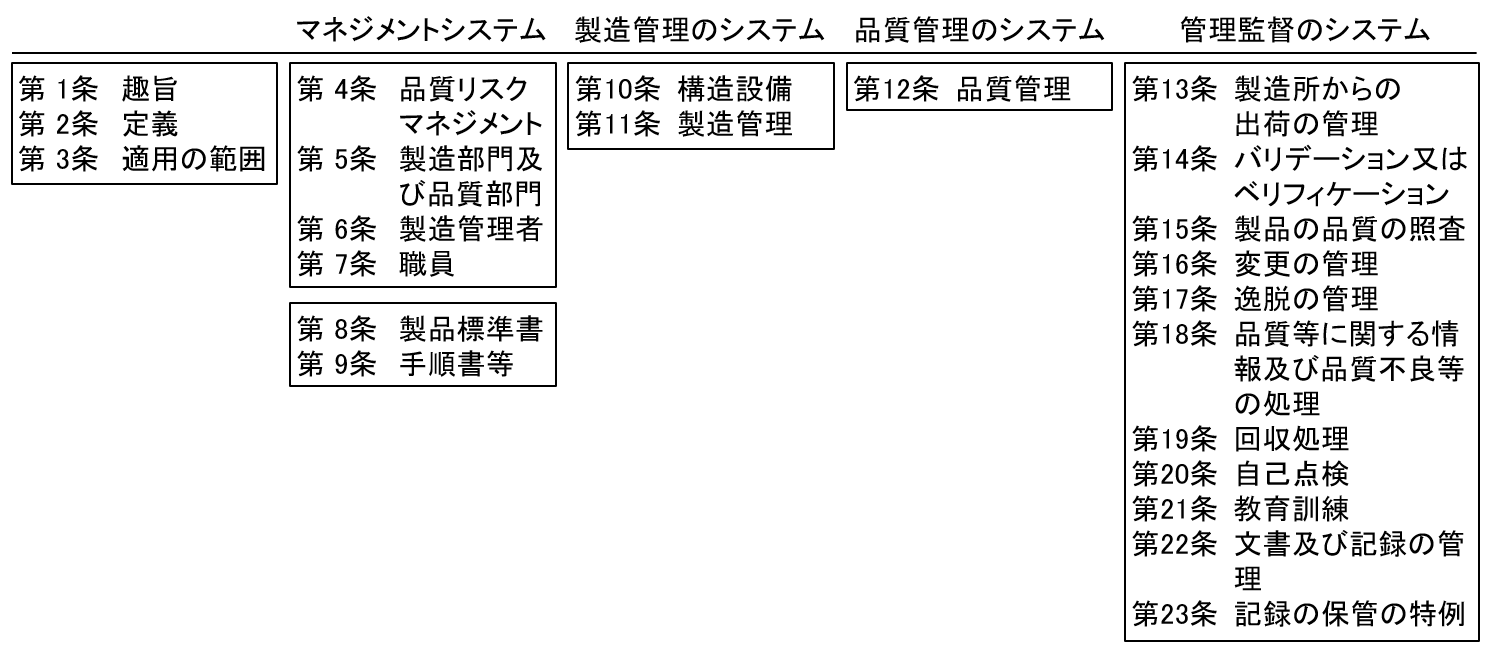
図. GCTP省令における条文の構成
ここまで、生きた細胞を製品とする細胞製造性を考慮した再生医療等製品の製品設計の考え方と、それに付随して生じる製品の同等性を担保する製造再現性の考え方について、また具体的なお話しとして、製品の品質特性(CQA)と切り離すことの難しい原料等や細胞加工の操作など、工程設計について雑感を述べました。今回からは、これらの考え方を前提とした、運用設計(マネジメントシステム)についてお話しをしたいと考えております。それに伴い、本稿では、GCTP省令の復習をあらためて行わせていただき、また、昨年11月に厚労省の事務連絡で発出された「再生医療等製品の無菌製造法指針」について雑感を述べさせていただきます。
● あらためて、GCTP省令に書かれている内容を確認する
GCTP省令については、第5回において、GMP省令との違いについて概説しました。今回は、再生医療等製品固有の考え方を除外し、「製造できること」を前提として、あらためて、GCTP省令について整理してみましょう。GMP省令を理解している方には常識の範疇となる内容ですが、お付き合いいただければ幸いです。
GCTP省令(平成26年厚生労働省令第93号)は、第1条から第23条までの文章で構成されていますが、その構成は前述した通り、GMP省令と変わりません。第1~3条(趣旨、定義、適用範囲)を除くと、図のように、品質確保において最も重要となる、品質リスクマネジメント(祭4条)、製造部門及び品質部門(第5条)、製造管理者(第6条)および職員(第7条)の要求が示され、品質リスクマネジメントを含む、マネジメントシステムの構築についての要求が前提となります。ここで特に求められるのは、トップマネージメントによる組織体制と、適切な職員の充当となります。その上で、品質確保すべき製品を対象とした製品標準書(第8条)と手順書等(第9条)の準備について、文書化要求が行われています。製品標準書には、製品規格や原料等の基準、製造方法および手順、品質の試験方法および手順、製品の包装形態、表示材料および表示方法、保管条件、細胞加工作業者に要求されるスキルなどを全て記載すべきものであり、再現性よく製造が実施できることを前提としています。また、手順書等では、上位文書として、衛生管理基準書、製造管理基準書、品質管理基準書(三基準書)を必須とし、後述する出荷管理、バリデーション、品質照査、変更管理、逸脱管理、回収処理、自己点検、教育訓練、記録管理などに関する手順作成が要求されており、製造管理者は品質リスクマネジメントを活用することで、製品のライフサイクルを通して適切に製造管理および品質管理を運用できるようにする組織の構築が必須となります。実際に、GCTP調査に係るサブシステムの1丁目1番地は「組織」であり、その他の一番地は全て手順書・記録書であり、ここをクリアしていなければGCTP省令への対応はあり得ません。
執筆者について
経歴 ※このプロフィールは掲載記事執筆時点での内容となります


コメント
/
/
/
コメント